
Sequence Surveyor Instructions for Use
Data Format
Sequence Surveyor reads SQLite databases representing a set of genomes aligned at the gene-level and optionally composed of multiple chromosomes. For the purpose of comparison between genes within different genomes, matching sets of genes (orthologs) are identified by an integer label (the ortholog_group_id).
The basic schema for the database file is as follows (elements in italics are descriptions of the column data and NOT part of the schema):
|
TABLE genome |
||
|
genome_id |
integer PRIMARY KEY |
Default ordering of the sequences. Should start with zero and continue in counting order (i.e. 0, 1, 2, ...). |
| name | text | name of the genome |
|
TABLE chromosome |
||
| chromosome_id | integer PRIMARY KEY | Numeric identifier for a chromosome. Should start with zero and continue in counting order (i.e. 0, 1, 2, ...). |
| name | text | name of the chromosome |
| genome_id | integer | genome_id of parent genome |
| length | integer | number of genes in the chromosome |
|
TABLE gene |
||
| gene_id | integer PRIMARY KEY | Numeric identifier for a gene. Should start with zero and continue in counting order (i.e. 0, 1, 2, ...). |
| name | text | name of the gene |
| description | text | textual description of the gene |
| chromosome_id | integer | chromosome_id of the parent chromosome |
| index_in_chromosome | integer | order of the gene within the chromosome |
| start | integer | genomic coordinate where the gene begins |
| end | integer | genomic coordinate where the gene ends |
| strand | integer | 0 (backward strand) or 1 (forward strand) |
| ortholog_group_id | integer | Numeric identifier for a set of orthologs. Should start with zero and continue in counting order (i.e. 0, 1, 2, ...). |
Additional data about the genes can be added to the gene table by adding a new column. Any additional information must be of a numeric form (one of "int", "integer", "tinyint", "smallint", "mediumint", "bigint","unsigned big int", "int2", "int8", "real", "double", "double precision", "float", "numeric", "decimal(10,5)", "boolean") and can then be used as a mapping to color individual genes in the tool (see "Customizing the Display").
This data format is extensible to other types of sequential data problems where you are comparing ordered sequences of data where there is a relationship between elements in different sequences, such as standard (base-level) alignments and aligned protein sequences or even something as different as rank data (see TextDNA). The sequences map to genomes, subsequences to chromosomes, and elements being compared map to genes. Groups of matching elements share the same ortholog_group_id value. However, any valid Sequence Surveyor dataset must, at a minimum, have the above fields.
Display Overview
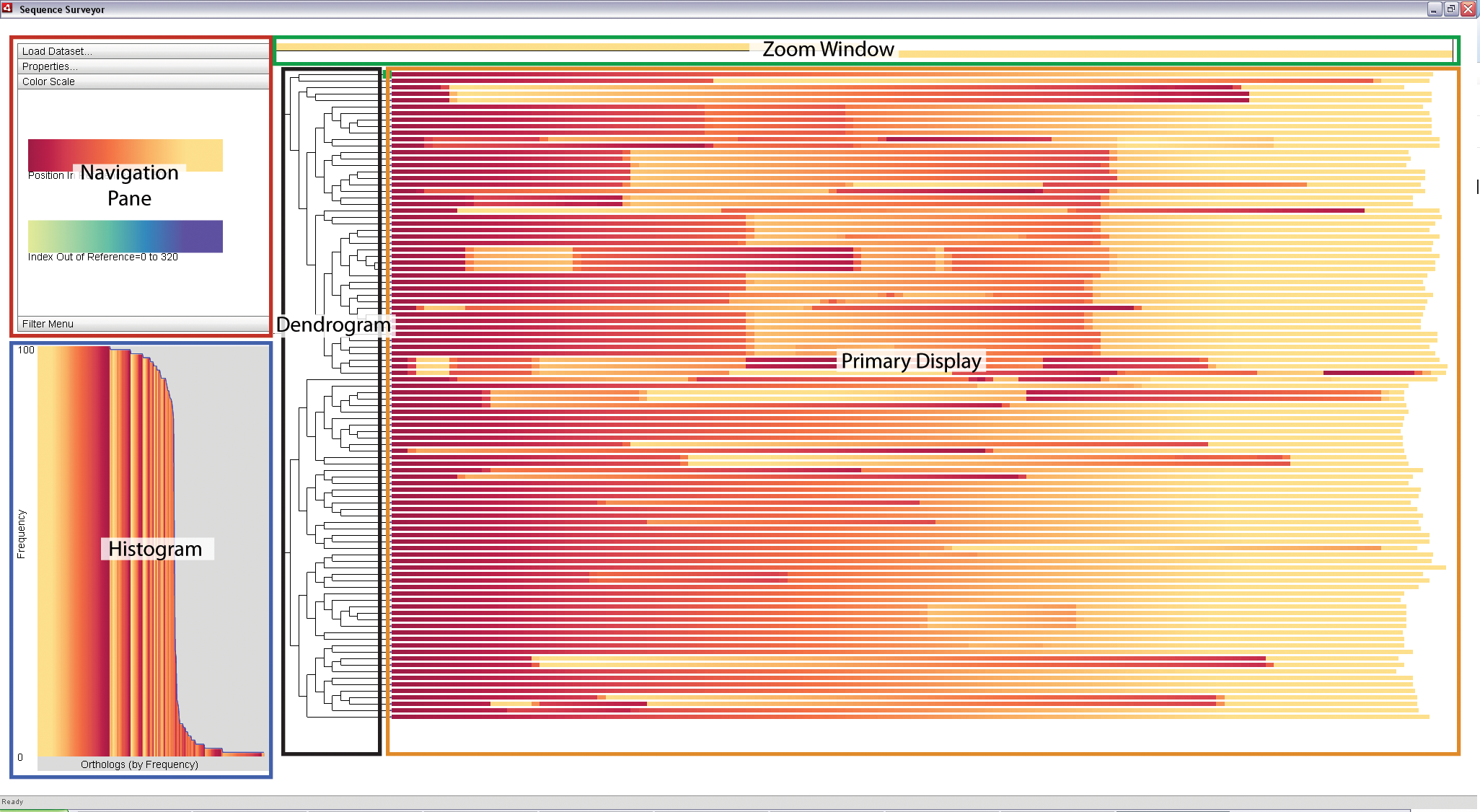
|
Sequence Surveyor leverages multiple visual displays to support visual analysis. The above image is the default Sequence Surveyor screen that appears when first loading a dataset. The components are as follows:
- Primary Display: An overview display of the data. Genomes map to rows; genes map to colors within the rows. If included, chromosomes are arranged from longest to shortest in the rows.
- Zoom Window: A detailed view of a specific blocked region within a row as selected by the user.
- Dendrogram: The phylogenetic tree (if provided) associated with the data.
- Histogram: A distribution of the matching sets of genes (ortholog groups) by frequency within the data (ortholog groups refer to sets of genes with same ortholog_group_id field (see "Data Format" for details)). Ortholog groups are sorted along the x-axis according to the number of genes in the ortholog group and colored with respect to the color parameters of the Primary Display. The height of a bar represents the average frequency of the ortholog groups represented by the bar; the blue line traces the maximal ortholog group size represented by the bar.
- Navigation Pane: This panel contains a variety of options for interacting with and exploring the data in the Primary Display. The components of this pane will be described in more detail in the section on "Customizing the Display".
Interacting with different elements of the display can affect other display components. For more detailed information on interaction with the various componenets of the display, see the ensuing sections of this guide.
Basic Use
To load a dataset into Sequence Surveyor, select the "Load Dataset" tab from the Navigation Pane. Click on the "Load" button to open the load file dialog box and navigate to the desired dataset on your computer (NOTE: files must have .db, .sql, or .sqlite extensions). Select the file and click "Open" to load the data into the tool. The phylogenetic tree is automatically loaded if the dendrogram's .tree file is of the same filename and same file location as the dataset (see"Customizing the Display" for more information).
The default display orders genome rows in the Primary Display by their position as specified in the .tree file if available and otherwise by their genome_id field (see "Data Format" for details). Rows can be rearranged by holding down the Alt key and clicking and dragging sequence rows vertically within the display. Crossing branches in the phylogenetic tree are then reduced in opacity. Within each row, neighboring genes are grouped into blocks, collections of genes aggregated spatially according to the Block Width parameter (see "Customizing the Display" for details). By default, each block is displayed as the average color of genes within the region represented by the block; however, alternative visual representations of the blocks are available.
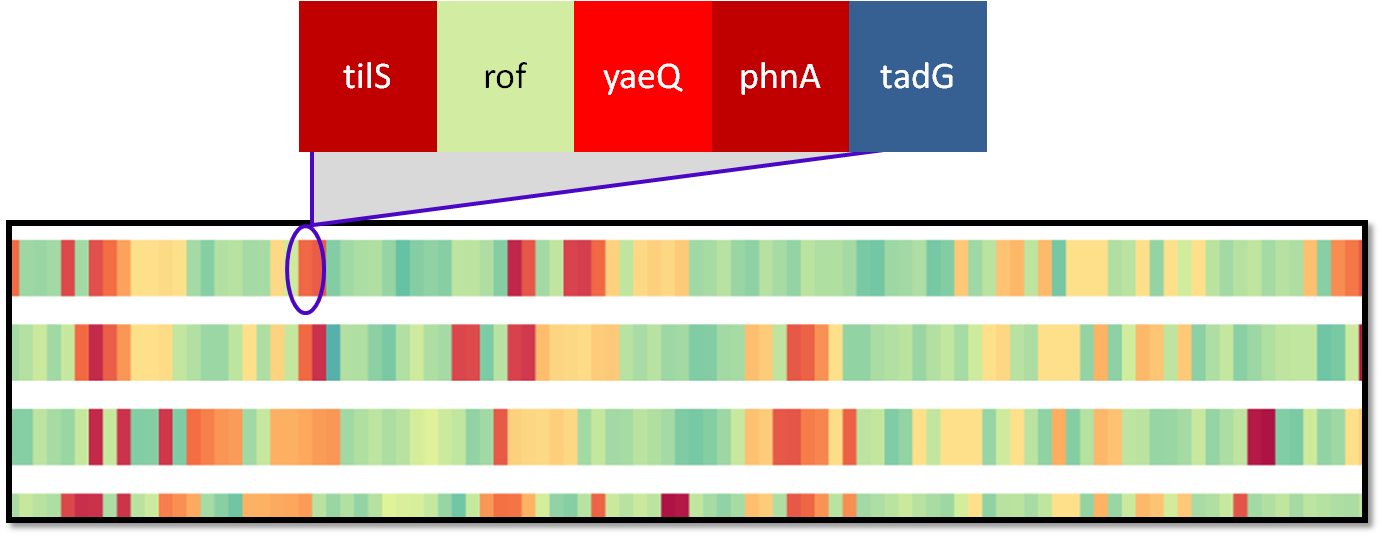 |
Genes within a particular spatial region are grouped into a single glyph, called a block. The width and representation of a block can be controlled by the user. |
Mousing over a block in the Primary Display will reveal a tooltip containing the names of the genes within the block, followed by the chromosome name and genome name on a separate line. If there are a large number of genes within a block, the list of names may be truncated, as indicated by an ellipse. Mousing over a block will also highlight any blocks in the Primary Display containing at least one gene orthologous to a gene in the current block, the locations of the component genes' ortholog groups in the Histogram display and the corresponding genes in the Zoom Window, if locked, as well as the active genome branch (green) and immediate sibling branchs of the tree (red). Clicking on a block in the Primary Display will cause lines to be drawn between all blocks containing at least one gene matching a gene in the clicked block. Links are only drawn between blocks sharing a matching gene. Clicking on the block again will remove the lines. Right clicking on any block and selecting "Clear Links" will remove all lines from the Primary Display.
Detailed information about the contents of a block can be accessed through the Primary Display. Right clicking on a block and selecting "Details" will open a pop-up containing a list of genomes in which genes orthologous to the component genes of the clicked block can be found. Selecting a genome name displays information about the orthologous gene in that sequence, including information from the description field of the database. Genes are indicated by '*' and blocks are separated by '***'. The blocks from the genome initially selected are preceded by "Target:" in the genome window.
Genes contained within a block can also be explored in the Zoom Window. Mousing over a block in the Primary Display will cause the components of the block to be displayed in the Zoom Window. Genes are displayed on either side of a horizontal guide line on the basis of their strand field to represent the forward and backward location of the genes on the original strand (see "Data Format" for details). The genes are represented as colored rectangles: their color and positioning correspond to their color and position values as defined by the parameters of the Primary Display. To interact with the components of the locked block, right click on the block in the Primary Display and select "Zoom to Block". The block is then locked into the Zoom Window and outlined in the Primary Display. If the component genes in the Zoom Window are dense, they may be aggregated into blocks, in which case, a zoomed block can be further explored by again locking the block in the Zoom Window.
Once locked, mousing over an gene will highlight blocks in the Primary Display containing at least one orthologous gene to the highlighted gene, the branches of the genomes in the Dendrogram for genomes containing orthologous elements to the highlighted gene up to the most recent common ancestor, and the location of the ortholog group in the Histogram distribution. Clicking on a block will draw a connecting link between blocks containing orthologous genes in the Primary Display. Clicking again will remove the link. Right click on any block in the Primary Display or Zoom Window and select "Unlock Zoomed Block" to release the block from the Zoom Window and return to mouseover zoom in the Primary Display.
Mousing over a bar in the Histogram display creates a tooltip with the list of ortholog_group_id values of the ortholog groups represented by the bars. It highlights branches of genomes containing genes from that ortholog group in the Dendrogram to the most recent common ancestor and the corresponding blocks containing the gene in the Primary Display. Clicking and dragging the mouse in the Histogram display draws a red rectangle in the Histogram. Releasing the mouse filters the data in the Primary Display: blocks containing genes corresponding to the bars within the rectangle remain fully opaque, while all other blocks are made partially transparent. Pressing escape removes this filter and clears the Histogram display.
Customizing the Display
The Navigation Pane contains a variety of menus for customizing the display. Selecting a tab in the Navigation Pane will open the corresponding menu set. This section will discuss these menus and how they can be used to support analysis in Sequence Surveyor. See the paper for details and examples of the types of analysis that these customizations can support.
Load Dataset
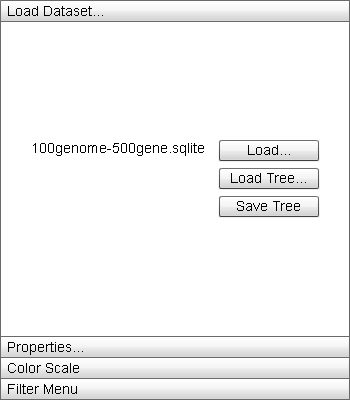 |
The "Load Dataset" menu contains options for interacting with and changing the underlying data of the display. |
- "Load" button: Opens a load file dialog box to load a new dataset into Sequence Surveyor. The new dataset will replace the currently visualized database. The dataset file name will appear to the left of the button.
- "Load Tree" button: Opens a file load dialog box to locate a .tree file to load into the database. If the file does not match the data, the default ordering (by ascending genome_id) is employed. The first line of the .tree file is the tree represented in the Newick format:
(A:0.1,B:0.2,(C:0.3,D:0.4)E:0.5)F;
Where the letters represent node names and the distance values and names of internal nodes are arbitrary. The names of the leaf nodes must match the genome names. Trees are assumed to be binary. The second line is a comma-delimited list of genome names representing the desired order of genome rows in the display from top to bottom. This line is optional. - "Save Tree" button: Opens a file save dialog box to save a .tree file containing the current Dendrogram structure and order of the genome rows in the display.
Properties
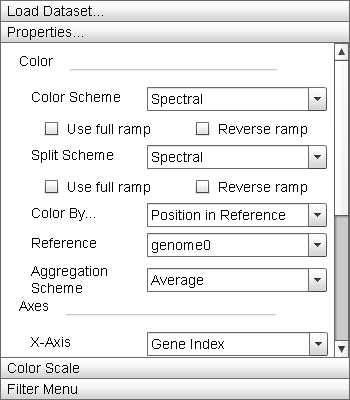 |
The "Properties" menu allows the user to manipulate the display parameters of the Primary Display. The "Update" button adjusts the parameters of the display to match those set in this menu. |
- "Color" Menu Section:
This menu section contains parameter settings for changing the element color mapping properties of the Primary Display, Zoom Window, and Histogram.
- "Color Scheme" dropdown list: This dropdown contains the actual color ramps that are mapped to genes. By default, Sequence Surveyor offers nine different color ramps and a grey ramp. Selecting the "Reverse ramp" checkbox below the dropdown list will invert the ramp before applying it to the data. Selecting the "Use full ramp" checkbox will cause the entire ramp to be used when using split color encodings.
- "Secondary Scheme" dropdown list: This dropdown is used to specify the secondary color ramps used when using split color encodings. If the same ramp is specified in both the "Color Scheme" and "Secondary Scheme" lists, split encodings will use the first half of the ramp as the "Color Scheme" and the second half as the "Secondary Scheme".
- "Color By" dropdown list: This list defines the property of a gene that is mapped to the gene color. There are six default property colorings. Additional colorings may be added by adding additional columns to the input database (see "Data Format" for details). Valid supplemental colorings are referenced by column name in this list. "Grouped Frequency Ordering" and "Position in Reference" are split coloring properties: a portion of the genes are mapped to one ramp while the remainder are mapped to another.
- Gene Index: Genes are colored according to their relative order within a genome. This coloring can be used to highlight positional patterns between genomes. (NOTE: Since this value is variable within ortholog groups, the Histogram is colored using a constant value.)
- Gene Frequency: Genes are colored according to the number of genes orthologous to it found within the dataset.
- Grouped Frequency Ordering: Orthologous genes are colored according to the set of genomes in which the genes are found. A total ordering is achieved by further sorting genes by their overall frequency (i.e. the number of genes in the ortholog group), their position in the genomes in decending order, and their ortholog_group_id value respectively. Orthologous gene sets with at least one gene in each genome are mapped to the "Color Scheme" ramp, while the remaining are mapped to the "Secondary Scheme" ramp. This property highlights patterns of co-occurence between orthologous sets of genes.
- Membership Frequency: Orthologous genes are colored according to the number of genomes they appear in. This is different from Gene Frequency coloring as it considers paralogs (a set of multiple orthologous genes within the same genome) as one orthologous element, where Gene Frequency considers each instance separately.
- Start: Genes are colored according to their start value (see "Data Format" for details). This represents the genomic coordinate where the gene begins. (NOTE: Since this value is variable within set of orthologous genes, the Histogram is colored using a constant value.)
- Position in Reference: Genes are colored according to the position of the first matching element in a selected reference genome (i.e. the Gene Index value for a particular ortholog within the reference genome. This genome can either be selected through the "Reference" dropdown list immediately below the "Color By" dropdown list or by right clicking on a target genome and selecting "Set as Color Reference". Genes orthologous to a gene in the reference genome are colored according to the "Color Scheme" ramp and remaining genes are colored according to the "Secondary Scheme" ramp, with colors being assigned according to the order in which an orthologous gene is first found in the remaining genomes, from top to bottom. This coloring highlights the movement of genes from one genome across the remaining genomes. The reference is indicated in the Dendrogram by a colored box placed before the genome row.
- "Aggregation Scheme" dropdown list: Sequence Surveyor offers four block encodings which display different statistical properties of the colors of the genes represented by the block.
- Average: Each block is represented as a solid color representing the average value of the "Color By" parameter across genes in the block.
- Color Weaving: Each block represents the approximate distribution of values of the "Color By" parameters across genes in the block. Each pixel is mapped to the value of one gene. After each gene is mapped to a pixel, the genes are randomized and mapped to the remaining pixels. The randomization repeats until the block is filled.
- Event Striping: This aggregation method highlights outliers and points of change within a block. Outliers (a data value greater than 1.5 times the inner quartile mean of values in the block) are mapped as stripes on the block. Dominant changes to the average value are visible as immediate shifts in the background color.
- Robust Average: Each block is represented as a solid color representing the average value of the "Color By" parameter across the genes in the block, excluding local outlier values.

TextDNA supports nine Color Brewer color schemes and a solid grey scheme.
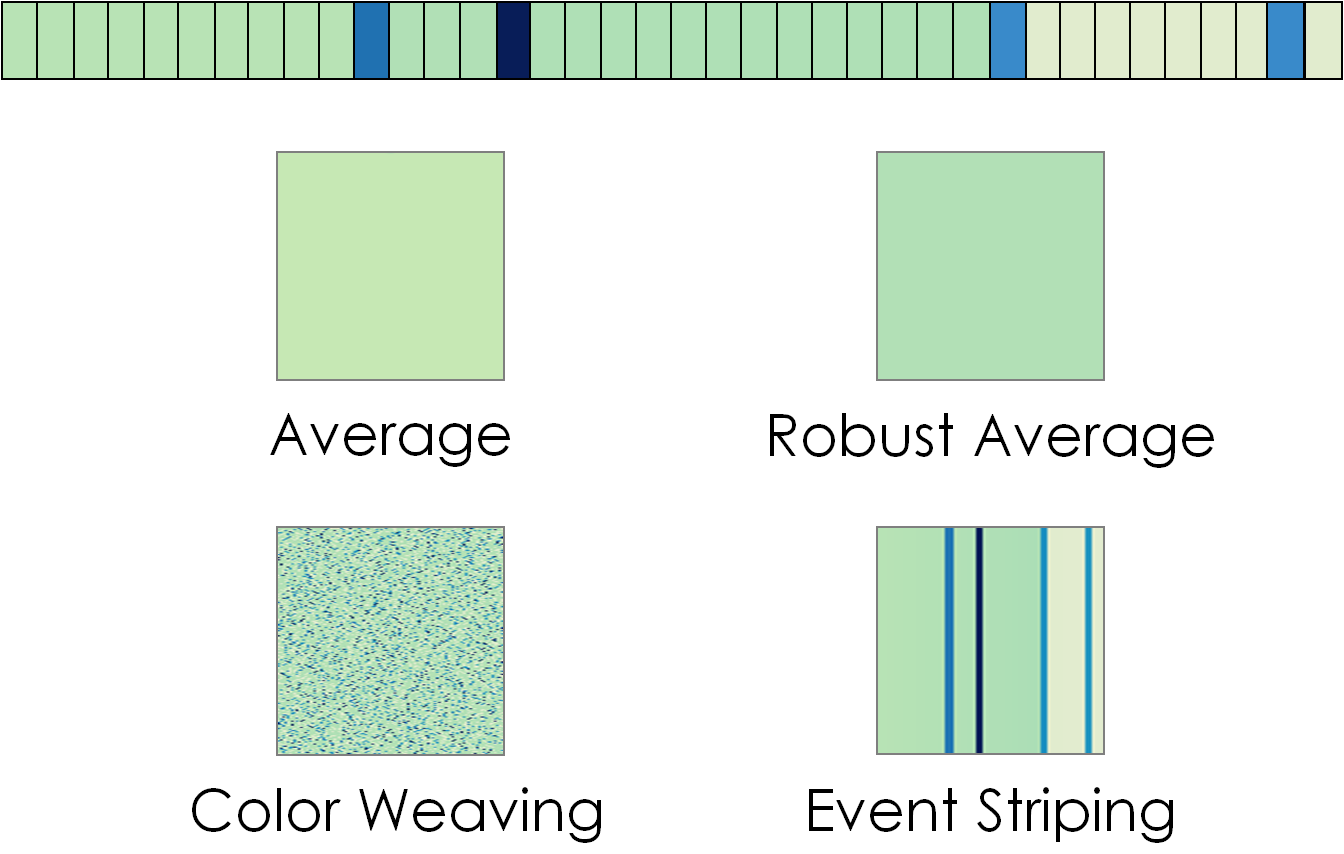
Sequence Surveyor supports four visual aggregation types. Here, those aggregate representations are applied to the top sequence of colored genes.
- "Axis" Menu Section: This menu section contains parameter settings for the ordering of genes within genome rows in the Primary Display and Zoom Window. Manipulating the ordering of genes within the genomes can highlight specific trends within a dataset by visually clustering like gene.
- "X-Axis" dropdown list: This list defines the setting for the x-axis ordering of genes within a genome. Sequence Surveyor provides four ordering options (Grouped Frequency Ordering, Start Position, Position in Reference, and Gene Index), all corresponding to their description in the "Color By" dropdown list. For position in reference ordering, the reference genome can be set using the "Reference" dropdown list immediately below the "X-Axis" dropdown list or by right-clicking on the target genome and selecting "Set as Position Reference".
- "Normalize block size" radio button: When selected, this box indicates that all genes are represented using the same normalized width. Otherwise, genes are represented using the difference between their start and end values, as specified by the database (see "Data Format" for details).
- "Glyph" Menu Section: This menu section contains the parameters that control spacing and sizing in the Primary Display.
- Block Grouping Width: This parameter defines the minimum width, in pixels, covered by a block. A larger block grouping width implies a greater number of genes will be aggregated into each block and vice versa.
- Glyph Height: This parameter defines how tall each block, an in turn, each genome will be. By default, this parameter is set to maximize the size of the genomes while keeping all genome rows visible.
- Vertical Space: This parameter defines how much white space is placed between each genome. By default, this parameter is set to be approximately half of the glyph height.
- "Update" button: This button applies the settings defined in the Properties menu to the visualization, updating the Primary Display, Histogram, and Zoom Window.
- "Reset" button: This button resets the Properties menu and visualization to the default parameters of the tool.
- "Cancel" button: This button resets the settings of the "Properties" menu to match those of the current Primary Display.
Color Scale
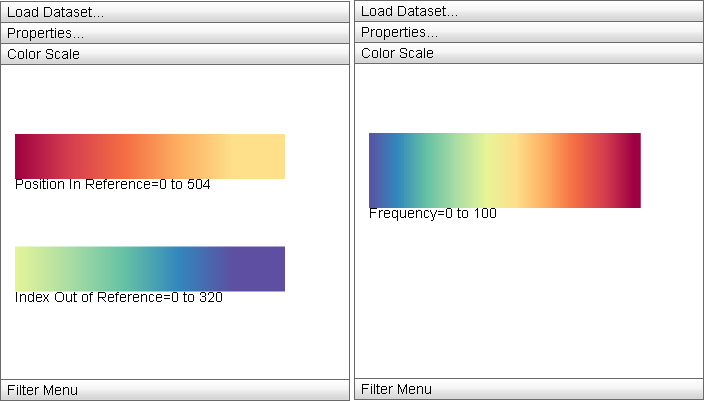 |
The "Color Scale" menu displays the currently active color mappings for the Primary Display. |
The Color Scale menu displays the detailed information about the current color mapping properties of the display: the active ramp is displayed from minimum property value to maximum and the minimum and maximum values are displayed below the ramp. For split color encodings, two ramps are used, one for the first portion of the data (that defined by the "Color Scheme" ramp) and the other for the second (defined by the "Secondary Scheme" ramp).
Filter Menu
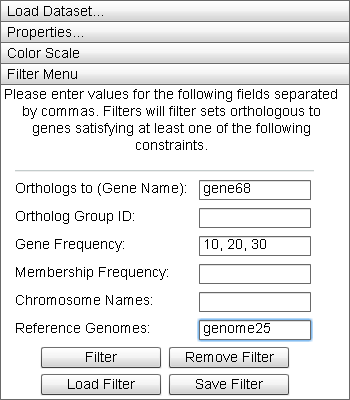 |
The "Filter Menu" allows the user visually filter the data displayed in the Primary Display. |
The Filter Menu allows the user to build conjunctive filters over the dataset. By entering data parameters in the text fields, a filter is constructed. Field data is applied as follows:
- Orthologs to (Gene Name): Filters for all genes orthologous to genes of the names in this list.
- Ortholog Group ID: Filters for all the genes whose ortholog_group_id field matches the values in this list.
- Gene Frequency: Filters for all genes orthologous to a certain quantity.
- Membership Frequency: Filters for all genes with orthologs in a certain number of genomes.
- Chromosome Names: Filters for all genes orthologous to at least one gene in the chromosome whose name is listed here.
- Reference Genomes: Filters for all genes orthologous to at least one gene in the genome whose name is listed here.
Clicking on "Remove Filter" will clear the current filter boxes and restore the opactity of all filtered blocks. The "Load Filter" button will open a load file dialog box. Opening a .filter file using this dialog will load the parameters of the filter into the "Filter Menu". The "Save Filter" button will open a save file dialog box to write out the filter parameters to a .filter file. All .filter files are of the format:
gene_names:
<comma-separated list of gene names>
gene_orthologs: <comma-separated list of ortholog_group_ids>
gene_frequencies: <comma-separated list of frequencies (ortholog group sizes)>
gene_membership_frequencies: <comma-separated list of membership frequencies>
chromosomes: <comma-separated list of chromsome names>
genomes: <comma-separated list of genome names>
Project Page | Sequence Surveyor | TextDNA
Adobe AIR must be installed to run Sequence Surveyor and TextDNA.
Email dalbers@cs.wisc.edu for more information.