Introduction
This page contains details about the LayerCake tool for visualizing viral population variability.
For a tutorial on how to interpret LayerCake, click here.
For downloads for the interactive LayerCake datasets associated with "High Genetic Diversity and Adaptive Potential of Two Simian Hemorrhagic Fever Viruses in a Wild Primate Population", click here.
For information about previous versions of LayerCake, including the paper "Visualizing Virus Population Variability From Next Generation Sequencing Data", click here
Contents
Tutorial
Overview
 |
| The entire LayerCake tool, on the krc1 dataset. |
The LayerCake tool is designed to allow viewers to quickly find areas of a viral genome where many different populations have variation with respect to a reference genome. There are two main components of the tool: rows, which represent distinct viral populations (in this case each row represents the viral population within each infected individual), and blocks, which represent a particular section of the genome. Since the information for the entire genome cannot fit in standard displays, each block is usually composed of the information for multiple individual base pairs averaged together.
 |
| The overview section of LayerCake |
The top-most component of LayerCake contains two items: a histogram of the average percentage of variant reads across all rows, and the location of ORFs in the reference genome. The more red a particular section of the histogram is, the more common variants are at that particular location. The color and resolution of the histogram changes as the user updates the parameters of LayerCake as a whole.
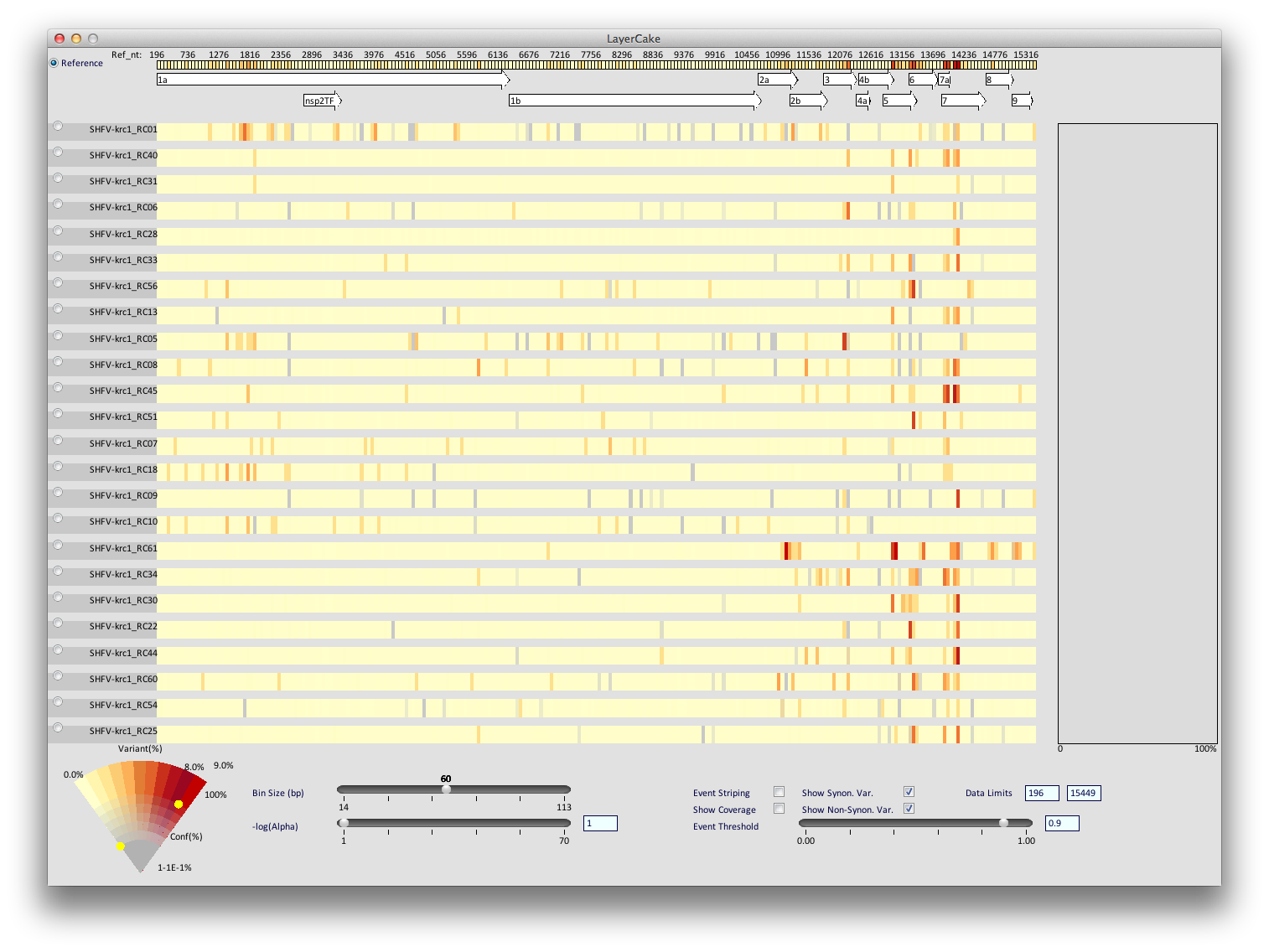
Color Wedge
  |
.png) .png) |
.png) .png) |
| The wedge legend/interaction tool. As the percentage of variant reads at a particular location goes up, that location becomes more and more red. As the certainty (in terms of data quality) of reads in a location goes down, the location becomes more and more greyed out. The yellow dots can be moved by dragging the mouse to control how the data are colored. This image shows how manipulating the upper dot might change the coloring of a particular row. |
In the bottom left is the wedge, which is both a legend for the coloring scheme used for LayerCake and also an interaction tool that allows the viewer to change this scheme on the fly. By dragging the two points of interaction, color can be used to dynamically determine regions of interest and make those regions visually salient. For instance one could grey out regions with variance that is too low to be considered (by moving the bottom yellow dot to the right), or where there is insufficient confidence in the data (by moving the bottom yellow dot upwards).
Finding Details
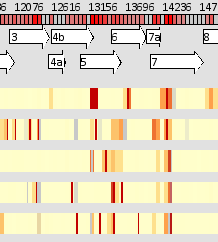
 | .png) |
| By mousing over a block, the space on the right is dedicated to showing the relative frequency of variants at that location across all rows. A tooltip gives more detailed information about coverage for a particular row. By right clicking on a selected block, the block expands, showing the variation information at the level of individual base pairs. Mousing over a location will show the relative proportion of different nucleotides in the reads across all rows. | |
Since by necessity LayerCake cannot show the variation information for every single basepair of a genome simultaneously on the same screen, regions of the genome are aggregated into blocks. By default each block contains information from 60 individual nucleootide locations, but this can be controlled by the user. Right clicking on a block causes it to expand outward, showing the detailed information of each nucleotide is presented by compressing the rest of the display. Mousing over a particular block, or a particular location of an expanded block, lets viewers compare behavior for every row at a glance.
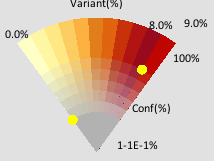
Another way of retrieving smaller-scale details from blocks is with "Event Striping." The viewer sets a threshold of interest (for instance, "locations where 50% or more of the reads are variants") and then LayerCake will display a red stripe at the location of each event which matches this criterion, obscuring some of the block to display this more salient information. The histogram at the top then becomes colored to reflect which columns have the most events across all rows.
 |
| An example of event striping for a section of the krc1 dataset, in this case showing events where 45% or more of the reads are variants. Since the blocks as a whole average together information from multiple locations, locations which are strong outliers that are dissimilar from their neighbors may be lost. Event striping allows viewers to recover this sort of information without having to investigate each block individually. |
Interactivity
 |
| The main controls for LayerCake |
The bottom of LayerCake contains sliders and input areas for controlling the parameters of the display. Here is a description of each one:
| Bin Size | Controls how many base pairs are averaged together to generate one block. The limits of this slider are to guarantee that there are not too many blocks to show at once, and that each block is not so big that it cannot be investigated in the window. |
| -log(Alpha) | Controls the lower limits of read quality we are willing to accept. The default is 1 (meaning we immediately grey out locations where the average reads have a p-value of more the 0.10), and the maximum is 70 (meaning we grey out read qualities where p is greater than 10E-70. |
| Event Striping/Event Threshold | As described above, the checkbox determines whether or not to enable event striping, where red stripes are drawn at locations where high percentages of variants are detected. The event threshold slider determines this level of interest. For instance if it is set to 0.90 then locations with more than 90% variation count as events. |
| Show Synon. Var./Show Non-Synon. Var. | Non-synonymous variants are variants where the resulting codon generates a different amino acid than the reference sequence. Synonymous variants do not change the resulting amino acid. By toggling each of these checkboxes you can determine which of these two types of variants "count" for the purpose of display and aggregation. If neither are selected, then no variation will be shown. |
| Show Coverage | Determines whether not to normalize the graphs which are displayed to show coverage information (see below). |
| Data Limits | Allows the viewer to examine only a subset of the data. If the data limits are adjusted, then there will be fewer basepairs to display, so more blocks can be shown on screen, and more resolution is possible. You cannot adjust the range of the data beyond the beginning of the first labelled ORF, or past the end of the last labelled ORF. |
 |
| An example of what a viewer might see after unchecking the "Show Coverage" checkbox: upon mousing over a region, the height of the bars reflects the read depth at a particular location. While this makes the proportion of variant reads more difficult to see, it provides another metric of data quality across the rows. Since not |
Manipulating Rows and References
By clicking and dragging the grey labels associated with each row, you can reorder the display order of rows in LayerCake. This allows the viewer to group and compare individual populations. Right clicking on the label will allow you to annotate it with one of four colors to further assist in labelling and reasoning about groups.
Clicking the radio box attached to each label will set that label as the new reference. By default, a read is counted as a variant if it conflicts with the consensus sequence for each individual. By choosing a particular population as a reference, it changes the definition of variant to mean the difference in read populations between one row and the new reference. e.g. if 50% of the reads at a location on the reference are "A" and 50% are "T", then a population of reads that is 100% A will be 50% different from the reference. This allows viewers to compare populations at a glance, and make claims about homogeneity of groups. For instance if there is a point that is very red in the default view, but remains red when a new reference is chosen, we can determine that not only are there variant reads at that location, but that those reads are highly idiosyncratic across a particular set of populations (since not only do they differ from the consensus sequence, they also differ from each other). To return to the default view, click the radio button next to the word "reference" in the upper left corner of the display.
 |
| An example of selecting a new reference. Here, the viral population of RC01 is the reference for determining variation. If the reads of particular individual are proportionally entirely the same as the new reference, there is no variation. If they are entirely different, then they are 100% variant. The chosen reference is identical to itself, so naturally it has no reported variation at any location. Places that are red across all of the remaining rows are idiosyncratic variants: the reference population differs from all other populations at these locations. |
Downloads
These stand-alone versions of LayerCake contain the datasets associated with the paper "High Genetic Diversity and Adaptive Potential of Two Simian Hemorrhagic Fever Viruses in a Wild Primate Population." Each is a zip file of approximately 18MB.
 |  |
| krc1 (Mac Version) | krc1 (Windows Version) |
| krc2 (Mac Version) | krc2 (Windows Version) |
LayerCake requires a Java Runtime Environment, which can be downloaded here.
This application was created on Windows, which does not
properly support setting files as "executable",
a necessity for applications on Mac OS X.
To fix this, use the Terminal on Mac OS X, and from this
directory, type the following:
chmod +x LayerCake.app/Contents/MacOS/JavaApplicationStub
Further questions and technical support can be directed to mcorrell@cs.wisc.edu
Past Versions
For the paper describing the initial system: Visualizing Virus Population Variability From Next Generation Sequencing Data
For an in-browser demo of the initial system (requires an in-browser Java Runtime Environment): LayerCake